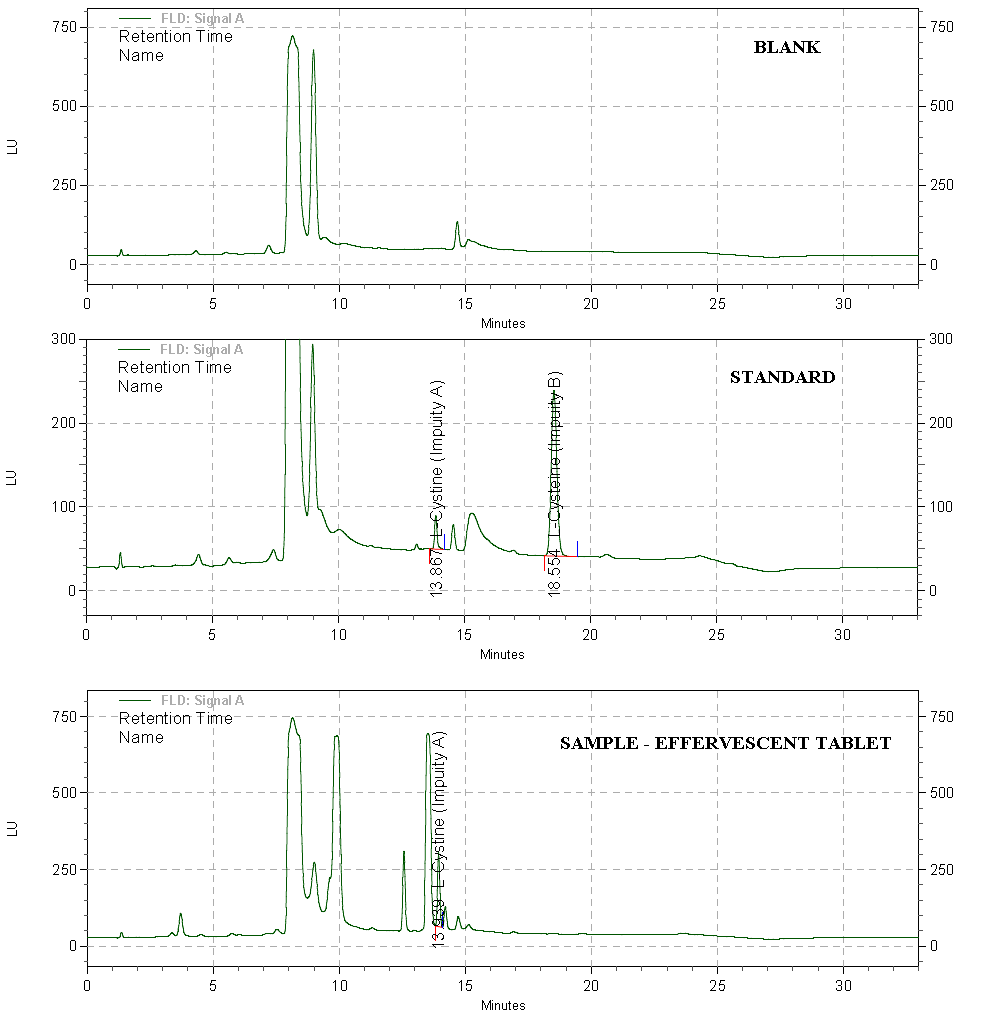
- Introduction
N-Acetylcysteine (acetylcysteine or n-acetyl-l-cysteine or NAC or (2R)-2-acetamido-3-sulfanylpropanoic acid) acts as a mucolytic agent and used to reduce the viscosity of pulmonary secretions in respiratory diseases. It is also used as an effective antidote in the treatmentof paracetamol poisoning [1-3].
The mucolytic action of NAC is probably due to its ability to decrease the viscosity of secretions by breaking the disulphide bonds of the protein network [4].
Additionally, NAC is acting as effective antioxidant and being studied for the treatment of various diseases such as nephropathy [5], liver failure [6], chronic obstructive pulmonary disease [7] and brain disorders [8]. It has also been used as a metal chelating agent [9]and radioprotective agent [10].
Liquid chromatography (LC) methods either as a conventional method or derivatization method have been